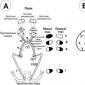
Distrofija (degeneracija) mrežnice je patološki proces, zaradi katerega je moteno trofično tkivo in poškodovane so celice mrežnice.
Mrežnica je notranja občutljiva očesna membrana, ki je sestavljena iz številnih fotosenzitivnih celic (palice in stožci). Zaznava svetlobne signale, izvaja primarno analizo in jih pretvarja v živčni impulz, ki vstopa v možgane.
Skotopični posnetki so bili izvedeni v temnih pogojih pod temno rdečo svetlobo na dveh ravneh osvetlitve, da bi razkrili največje kombinirane odzive palice in stožca. Podatke smo preverili za normalno s Kolomogorov-Smirnovim testom. Vsi podatki niso pokazali odstopanja od normalne porazdelitve.
Kaj je distinalna distrofija
Številne dedne bolezni ledvic imajo diagnostične funkcije mrežnice. Diagnoza dedne ledvične bolezni je pomembna zaradi tveganja nadaljnjih ledvičnih in sistemskih zapletov, posledic za druge družinske člane, predvidljivosti kliničnega poteka in možnosti zdravljenja.
Bolezen je v našem času precej pogosta. Ta patologija pogosto prizadene starejše in otroke.
Vrste distrofije mrežnice

- Dedno (primarno):
- generalizirana: pigmentirana in točkovna bela degeneracija mrežnice, prirojena amaurosis Leber itd.;
- osrednji (oslabljen osrednji vid): Stargardtova bolezen, Besta, senilna makularna degeneracija itd .;
- periferna (motnja lateralnega vida): X-kromosomska mladostna retinoschisis, Wagnerjeva bolezen, Goldman-Favrejeva bolezen itd.
- Pridobljeno (sekundarno).
Sekundarna distrofija se razvije v ozadju vpliva na mrežnico kakršnih koli zunanjih ali notranjih dejavnikov.
Poleg tega lahko abnormalnosti mrežnice pomagajo razložiti patogenezo bolezni ledvic in jo lahko včasih uporabimo za spremljanje njenega poteka. Podedovana bolezen ledvic, ki je prisotna pri odraslih in otrocih. Dedna bolezen ledvic se domnevno pojavi v družinski anamnezi s podobnimi simptomi ali ko anomalije prizadenejo več sistemov brez druge očitne razlage. Znane izjeme, ki temeljijo na trenutnem znanju, so avtosomno dominantne in recesivne oblike policistične ledvične bolezni, mišične cistične ledvične bolezni in subtilne nefropatije bazalne membrane.
Predisponirajoči dejavniki
- Intoksikacija.
- Okužbe.
- Bolezni krvnega obtoka.
- Sistemske bolezni vezivnega tkiva.
- Endokrina patologija, vključno s sladkorno boleznijo.
- Bolezni srca in krvnih žil.
- Presnovne motnje.
- Kajenje
- Poškodbe oči.
- Bolezen oči (kratkovidnost itd.).
- Nosečnost
- Debelost.
Klinične manifestacije bolezni

Podedovana bolezen ledvic, povezana z izgubo sluha. Podedovana bolezen ledvic pogosto vključuje tudi mrežnico, ker se ledvica in mrežnica razvijeta v isti fazi zarodka in delita razvojne poti; pregrada glomerularne filtracije in retinohoidni prehod strukturno podobna; glomerulus in horioretin sta velika kapilarna sloja in epitelijske epitelne in epitelne celice retinalnega pigmenta, ki so kritično odvisni od delovanja cilij.
Načini razvoja ledvic in oči
Epitelne celice imajo organsko specifične funkcije, osnovne membrane podpirajo sosednje strukture in tvorijo oviro, ki preprečuje prehajanje makromolekul, kapilare pa zagotavljajo prehrano in odstranjujejo odpadke iz bližnjih metabolično aktivnih celic.
- Poslabšanje vizije somraka.
- Fotofobija
- Zmanjšan osrednji vid.
- Izkrivljanje oblike predmetov.
- Pojav bliskavic, muhe pred očmi.
- Zmanjšana ostrina vida.
Retinalna degeneracija je progresivna bolezen in vodi do postopnega zmanjševanja vida in slepote. Lahko se kombinira z nistagmom, strabizmom. Pogosto ima asimptomatski potek. Najbolj neugodne v smislu prognoze so dedna degeneracija mrežnice.
Mutacije, ki vplivajo na beljakovine v podocitih cilij ali sorodnih strukturah, vodijo do cistične bolezni ledvic, vključno z nefronofizo in njenimi različicami ter Bardé-Bidelovim sindromom. Retina je običajno prizadeta, druge klinične značilnosti pa so izguba sluha, nenormalen razvoj okončin in številk, razvojna zakasnitev, sinusna inverzna bolezen, bolezni jeter in dihal ter neplodnost.
Nenormalnosti pilinga pri dedni bolezni ledvic vključujejo kolobom, peritoneum, atrofijo in pigmentacijo, hamartome, žilne anomalije in kristale. Te spremembe lahko spremljajo tudi retikularne povezave ledvične odpovedi, kot so krvavitev, zoženje arteriole, eksudati, srčni napadi, kalcifikacija in makularna degeneracija.
Razmislite o najpogostejših možnostih za distrofijo mrežnice.
Retinalna pigmentna distrofija
Gre za dedno patologijo mrežnice, pri kateri so predvsem palice prizadete. Nekaj let pred pojavom sprememb v fundusu pri bolnikih s temno prilagoditvijo je motena. Glavne manifestacije bolezni so hemeralopija (nočna slepota) in zoženje vidnega polja. Lezija je dvostranska, za katero je značilno odlaganje pigmenta v fundusu, medtem ko se žile zožijo, razvije se uničenje optičnih vlaken. Slepost je praviloma 40 let.
Podedovana bolezen ledvic, povezana z motnjami mrežnice. Bolezni mrežnice pri dedni bolezni ledvic. Optična jama. Vid je bil v tem očesu normalen, koloboma pa je običajno asimetrična. Anomalija jutranje slave. Vid je bil zlomljen. Normalna osrednja mrežnica. Osrednja mrežnica je bila normalna tudi v očeh drugih. To je periferna mrežnica obraza, katere dno je prikazano. Kolostomija mrežnice je bila prisotna na obrobju na dvostranski osnovi. Retinopatija razbremeni makulo in je dvostranska.
Vizija ni bila prizadeta; Periferna retinopatija pri Alportovem sindromu. Ponovno, vizija ni prizadeta. Široko razširjeni veliki mehki makularni možgani v membranoproliferativnem glomerulonefritisu. Pozna oblika gostih ledvičnih obolenj z obsežnim ledvičnim izpiranjem mleka, krvavitvijo in atrofijo. Te osebe je treba oceniti in spremljati oftalmolog zaradi tveganja zapletov in možnosti zdravljenja. Atrofija mrežnice pri Joubertovem sindromu, druga oblika nefronofov, dokazana z optično koherentno tomografijo.
Prirojena amaurosis Leber
Bolezen se manifestira od rojstva in ima hud potek. Otroci nimajo osrednjega vida, bodisi so slepi ali pa izgubijo vid pred starostjo 10 let. Pregled je pokazal strabizem, nistagmus, različne nevrološke simptome, izgubo sluha in nevropsihično razvojno zakasnitev. Pri pregledu se v očesnem bazu odkrijejo pigmentne usedline, glava svetlega živčnega živca.
Bolnik je sprva imel slab vid, ki je napredoval. Atrofija korio-retine s sindromom Kearns Syrah. Retina je razredčena in depigmentirana. Vizija je bila prizadeta. Hamartoma pri tubularni sklerozi. Bela gomolja s tankim linearnim krvavitvami so bila enostransko prisotna. Ta nenormalnost se pojavi pri večini bolnikov z gomoljno sklerozo in se mora razlikovati od infarkta mrežnice zaradi hipertenzije, sladkorne bolezni ali lupusa. Corkscrew plovila v Fabry sindrom.
Točkovna distrofija
To je asimptomatsko, vendar se lahko z okluzijo osrednje arterije pojavi nenadna izguba vida. Razlikovati jih je treba od sprememb v hipertenziji. Von Hippelov sindrom Lindau. Opazili so želodčne žile, ki so vodile do hemangioblastoma tik izven polja. Očitni makularni lipidni izločki so posledica puščanja iz nenormalnih žil. Te napake se običajno nahajajo v spodnjem delu šarenice, horioretina ali optičnega diska. Kolobom optičnega diska najdemo pri vezikoureteralnem refluksu in drugih strukturnih boleznih sečil.
Spot bela distrofija mrežnice
Bolezen se razvija v otroštvu, ima družinski značaj, počasi napreduje. Pri bolnikih z mračno in nočno slepoto, zoženo vidno polje. Na fundusu so bile razkrite belkaste lezije, znaki atrofije vida.
Sindrom ledvičnega koloboma
Koloboma je običajno asimetrična, na primer z normalnim očesom ali šibko okvaro na enem očesu in z resno centralno nepravilnostjo v drugem. Podobno se resnost okvare razlikuje med posameznimi družinskimi člani. Zdravljenja ni, zato so lahko zapleteni zaradi glavkoma, odcepitve mrežnice in osrednje serozne retinopatije. Devetdeset odstotkov otrok ima kolobom optičnega diska, horioretin ali šarenico, 20–40% ima strukturno anomalijo sečil, vključno z solitarno ledvico, ledvično hipoplazijo, duplex ledvicami ali vezikoureteralnim refluksom, 60% pa ima srčne napake.
Stargardtova bolezen (makularna distrofija)
Ta patologija se manifestira pri starosti 3-5 let, obremenjena dednost pa predispozira za njen razvoj. Otroci se pritožujejo zaradi zamegljenega vida, fotofobije. Po nekaj letih po pojavu bolezni izginjajo osrednja vidna polja, ostrina vida se hitro zmanjšuje. Pregled je pokazal spremembe v pigmentnem epiteliju in atrofiji vidnega živca.
Težave z ledvicami so pogosto povezane z ipsilateralno paralizo obraza. Izguba teh genov povzroča okularne in urogenitalne nepravilnosti. Diafragma je nezadostna, optični živec in hipoplastična jama. Te nenormalnosti so lahko zapletene zaradi krhke roženice, glavkoma in odcepitve mrežnice. Pri 50% bolnikov pride do raka ledvic.
Distrofične bolezni mrežnice
Verjetno gre za podtip Joubertovega sindroma z boleznimi jeter. Bolniki imajo lahko znak molarnega zoba, malformacijo srednjega možganja in zadnjih možganov, ki so opazili tudi pri Joubertovem sindromu in prirojeni fibrozi jeter. Čeprav imajo medularne ledvične ciste, ne razvijejo nujno ledvične odpovedi. Coloboma optičnega živca, duševna zaostalost, nistagmus in prirojena ataksija.
Najboljša bolezen
Osnova te patologije je makularna degeneracija. Razvija se v otroštvu in adolescenci in zaznamuje zmanjšanje ostrine vida. Ko je oftalmoskopija odkrila dvostransko lezijo na licu mesta v obliki rumenega ognjišča.
Senilna degeneracija makule
Razvija se praviloma pri osebah, starejših od 50 let. Trpi predvsem ženski spol. Proces degeneracije pogosto poteka neopaženo. Bolezen se začne z poslabšanjem vida v eni ali dveh očesih in se obravnava kot prezbiopija. Z napredovanjem degenerativnega procesa pride do popolnega izginotja vida.
Alportov sindrom
Očitne so z oftalmoskopijo fundusa in fotografijami, zlasti brez rdečih slik. Drusen z gostim nanosom je velik in mehak ter se nahaja na makuli. Osebe z ledvično boleznijo in atrofijo mrežnice se najprej pritožujejo zaradi slabega nočnega vida in celo predornega vida. Na začetku se anomalija manifestira samo s pomočjo elektroretinograma, kasneje pa je videz mrežnice nenormalen s bledico, pigmentacijo in madeži, oslabljenimi arteriolami in venulami ter očitnimi žilnimi žilami.
Dedovanje je avtosomno recesivno. Redke motnje, povezane z recesivnimi mutacijami v genih, ki kodirajo druge proteine v cilijah, bazalnih telesih ali centrosomih epitelijskih celic ledvic in mrežnice. Včasih mutacije vplivajo na dva različna gena, resnost bolezni pa je lahko odvisna tudi od modifikacijskega gena. Za ta sindrom so značilne klinično žariščna in segmentna glomerularna skleroza, debelost, oslabljena kognicija in polidaktilnost. Fenotip se razlikuje v družinah, posamezniki pa morda niso prisotni do petega desetletja.
X kromosomski juvenilni retinoshizo
To je dedna distrofija retine, za katero je značilno razcepljenost. Pogosteje je pri moških. Otroci imajo zmanjšan vid, mežikanje. Pregled razkriva velikanske retinalne ciste, obdane s pigmentom, ki se lahko pokvari.
Wagnerjeva bolezen
To je dedna bolezen, katere izrazna značilnost je visoka stopnja kratkovidnosti. Pogosto imajo bolniki z 10–20 let motnosti leče in koncentrično zoženje vidnega polja. Bolezen je lahko zapletena zaradi glavkoma ali odcepitve mrežnice.
Retina je normalno zgodaj, vendar se kasneje pojavi atrofija brez velike pigmentacije. Nenormalnosti vida vključujejo nenormalno elektroretinogram, nočno slepoto, okvare perifernih polj in slepoto v zgodnji odraslosti. Nosilci imajo verjetno subtilne napake v funkciji mrežnice.
Druge klinične značilnosti so: jetrna fibroza, polidaktilija, cerebelarna ataksija in nenormalni gibi oči, vključno z rotacijskim nistagmusom, psihomotorično retardacijo in razvojno zakasnitvijo. Nenormalnosti luskavosti vključujejo retinitis pigmentozo in morebiti horioretinalni kolobom. Kot pri sindromu Elder Loken je retinitis pigmentosa pogosto huda, elektroretinogram pa je v zgodnji fazi nenormalen.
Bolezen Goldman-Favre
V tej patologiji je pigmentna distrofija kombinirana s steklasto lezijo, retinoshizo. Manifestacije bolezni so zamegljen vid, nočna slepota in oslabljena temna adaptacija. Pri bolnikih lahko zaznate izgubo vidnega polja v obliki obroča ali koncentrično zoženje.
Prišlo je do ledvične odpovedi, slabega sluha in zmanjšanega mišičnega tonusa, vendar ni nobenih nepravilnosti mrežnice. Meckel-Gruberjev sindrom lahko povzročijo mutacije v istih genih kot Joubertov sindrom, vendar je v zgodnjem življenju usoden. Zanj je značilna cistična displazija ledvic, jetrna fibroza, okcipitalni encefaloklorid in polidaktil.
Za Junov sindrom so značilne cistične bolezni ledvic, dolge ozke prsi, kratke limbične, brahidaktilne in druge okostje skeletov. Bolezen dihal je lahko huda. Prihaja do atrofije mrežnice, vendar blage. Imajo tudi izgubo sluha, moteno delovanje jeter, napredovalo kardiomiopatijo, zakasnjeno puberteto, debelost in sladkorno bolezen brez duševne zaostalosti, polidaktije ali hipogonadizma. Atrofija mrežnice se pojavi z velikimi pigmentnimi grudicami, vaskularnim slabljenjem in sijajem optičnega diska.
Diagnostika
 Oftalmoskopija omogoča zdravniku pregled osnove očesa.
Oftalmoskopija omogoča zdravniku pregled osnove očesa. Diagnoza distrofije mrežnice temelji na kliničnih manifestacijah, zgodovini bolezni, podatkih o pregledu in pregledu pri oftalmologu. Bolniki so:
Alagilov sindrom
Predstavitev poteka od otroštva do zgodnje zrelosti. Pogoste so tudi izgube sluha, kardiomiopatija in sladkorna bolezen, vendar se klinične značilnosti razlikujejo med družinami. Atrofija mrežnice se pojavi pri vsaj polovici bolnikov, elektroretinogram pa je 90% nenormalen.
Kearns-Sayrejev sindrom
Začetek je običajno do 20 let, včasih pa kasneje. Kearns-Sayrejev sindrom je povezan s poškodbo tubularnih tubularnih žil, pa tudi s kronično progresivno zunanjo oftalmoplegijo, ptozo, izgubo sluha, nenormalnostmi srčnega prevajanja, cerebelarno ataksijo in sladkorno boleznijo. Kljub atrofiji in pigmentaciji mrežnice je izguba vida običajno blaga.
- splošni klinični pregled (kri, urin, biokemija);
- oftalmoskopija (omogoča pregled fundusa);
- preiskava intraokularnega tlaka;
- fluoresceinska angiografija mrežnice (študija mrežničnih žil);
- ehooftalmografija (metoda proučevanja struktur zrkla z ultrazvokom);
- entoptometrija (omogoča vam popolno sliko o funkcionalnem stanju mrežnice).
Zdravljenje
Terapija za distrofne procese v mrežnici mora imeti celosten pristop. Njegov glavni cilj je čim bolj ustaviti napredovanje bolezni in izboljšati vid. Če je patološki proces, ki vpliva na mrežnico, sekundarni, je treba najprej zdraviti bolezen, ki je privedla do nje.
Med te motnje spadajo nevrofibromatoza, tuberkularna skleroza, von Hippelova Lindauova bolezen in Sturge-Weberjev sindrom. Von Hippelovi sindromi Lindaua in Sturge-Weberja prav tako povzročajo žilne anomalije in so obravnavane kasneje. Hipertenzija nastopi zaradi proksimalne stenoze ledvične arterije, koarktacije aorte ali femohromocitoma.
Vrste tuberkulozne skleroze 1 in 2
Nenormalnosti pilinga vključujejo ishemijo, hamartom, vključno s kombinacijo mrežničnega hamarmota in retinalnega epitelija mrežnice, optičnim gliomom in atrofijo optičnega živca, ki je posledica pritiska na vidni živec. 20% bolnikov nima mutacije. To vodi v hematurijo, včasih pa tudi do raka. 30% bolnikov ima ledvične ciste. Okular koloboma in tumorji vek so redki. Večina bolnikov ima vsaj eno gammo mrežnice ali optičnega živca.
Zdravljenje z zdravili
- Metabolični steroidi (nerobol, retabolil).
- Antikoagulanti (heparin, fraksiparin).
- Disagregant (aspirin, klopidogrel).
- Vazodilatatorji (nikotinska kislina, drotaverin, komplamin).
- Zdravila, ki izboljšujejo mikrocirkulacijo (pentoksifilin, Cavinton).
- Vitamini (A, B, C, E).
- Emoksipin, taufon v obliki kapljic za oko.
Zdravljenje daje boljše rezultate v zgodnjih fazah bolezni. Zdravila se uporabljajo za lokalno in splošno zdravljenje.
Kirurško zdravljenje
Z neučinkovitostjo konzervativnega zdravljenja, z namenom izboljšanja krvnega obtoka in prehrane tkiv, poteka revaskularizacija mrežnice. Uporablja se presajanje mišičnih vlaken iz rektusnih mišic očesa v perihondralni prostor.
Nekatere rezultate pri zdravljenju lahko dosežemo po foto-in laserski koagulaciji. Hkrati je vpliv usmerjen na poškodovana tkiva, ki so ločena od zdravih, kar preprečuje širjenje procesa in preprečuje odmik mrežnice.
Zaključek
Razvoj distrofičnih procesov v mrežnici očesa ogroža bolnika s popolno izgubo vida brez možnosti njegovega okrevanja. To se ponavadi pojavi v poznih fazah bolezni, če ni ustreznega zdravljenja. Bolezen bistveno zmanjša kakovost življenja bolnikov, slepota pa vodi do invalidnosti. V prisotnosti te patologije je treba zdravljenje začeti čim prej, ko se še niso pojavile nepovratne spremembe. To omogoča ohranitev vida in prekinitev bolezni.
NTV, program "Brez recepta", vprašanje "degeneracije mrežnice":
TV kanal "CTV.BY", program "Zdravo, doktor", vprašanje "Distrofija mrežnice - kaj je to":
Zahvaljujoč razvoju visoke tehnologije, je trenutno večina očesnih bolezni mogoče zdraviti. Vendar pa v oftalmološki praksi še vedno obstajajo patologije, s katerimi je zelo težko, včasih nemogoče obvladati. Pri tako hudih boleznih je mogoče z dobrim razlogom pripisati pigmentno distrofijo mrežnice. Posebnost te bolezni je dolgotrajen progresivni potek, ki se izmenjuje z obdobji ostrih vidnih motenj in nepričakovanimi remisijami. Bolezen se praviloma začne aktivno razvijati v adolescenci, do 20. leta starosti pa jo je že mogoče jasno diagnosticirati, do 40-50 let pa se ponavadi pojavi slepota.
Simptomi
Tipičen simptom te patologije je huda prizadetost vida, zlasti pri prehodu z normalne svetlobe na šibkejšo svetlobo, na primer v mraku. In v temi je »nočna slepota«. Znanstveno, ta pogoj se imenuje hemeralopija. Najprej se občutno zmanjša periferni vid, nato se vidni živci s časom atrofirajo, osrednji vid pa se koncentrično zoži. To je že znanilec popolne slepote.
Značilnosti bolezni
Pigmentirana distrofija (degeneracija, retinitis) je dobila ime zaradi značilnih sprememb v fundusu. Na mrežnici vzdolž mrežnice (na nivoju lokacije živčnih vlaken) so nastale posode temno rjave barve. To so svojevrstna kostna telesa različnih oblik in velikosti, ki so podobna kupu razsutega premoga. Sčasoma pride do razbarvanja celic pigmentnega epitela. Rezultat je, da se očesno bazo preliva in postane kot mozaik, ki nastane z gosto prepletanjem oranžno-rdečih koroidnih žil, ki se konvergirajo v središče očesa.
Z napredovanjem bolezni se poveča število organov pigmentnih kosti, razširi se območje razdeljevanja. Postopoma konglomerati pigmentnih mas gosto dotaknejo celotno mrežnico. Nato distrofične spremembe zajamejo osrednji del očesa. Retinalne žile se močno zožijo, postanejo filiformne, glava optičnega živca pa dobi voskasto bledo barvo z naknadno atrofijo. V obeh očesih se pogosto razvije pigmentirana distrofija, v redkih primerih pa lahko prizadene eno oko.
Vzroki razvoja
Izvor (etiologija) pigmentne distrofije še ni bil raziskan. Oftalmologi izražajo nasprotujoča si mnenja. Nekateri verjamejo, da je vzrok te patologije hipofunkcija dodatka možganov. Druge pogosto vplivajo na toksične poškodbe telesa ali na poliglandularno motnjo. Tudi drugi trdijo, da je razvoj bolezni posledica beriberija, endokrinih ali genetskih motenj. Hipoteza je dokaj stabilna, da presežek svetlobe osvetljuje posebno snov v organih vida - rodopsin. Posledica je, da je funkcija posodabljanja fotoreceptorjev mrežnice otežena. V tem primeru se vsi strokovnjaki strinjajo, da je glavni vzrok bolezni družinsko dedna.
Zdravljenje
Pigmentarno distrofijo diagnosticiramo z elektroretinografskimi raziskavami. Na žalost je napoved slaba. Patološki proces se lahko le upočasni. Glavna smer zdravljenja je izboljšanje trofizma mrežnice, ekspanzija žil, vključno z žilnimi. V ta namen uporabljamo nikotinsko kislino, acetilholin in pripravke na osnovi heparina. Na splošno je seznam zdravil, ki upočasnjujejo napredovanje pigmentne distrofije, precej obsežen. Zdravniki uporabljajo ščitnice, testise, jajčna zdravila, presnovne steroide (na primer Nerobolil, Retabolil), predpisujejo vitaminsko terapijo (razen vitamina A).
Informacije na naši spletni strani so informativne in izobraževalne. Vendar pa te informacije nikakor niso koristi za samozdravljenje. Posvetujte se z zdravnikom.
- Vkontakte 0
- Google+ 0
- Ok 0
- Facebook 0