Dystrofia (degenerarea) retinei este un proces patologic, ca rezultat al distrugerii țesutului trofic și a deteriorării celulelor retinei.
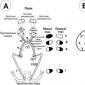
Retina este o membrană interioară sensibilă a ochiului, care constă dintr-un număr de celule fotosensibile (tije și conuri). Ea percepe semnalele luminoase, efectuează analiza primară și le transformă într-un impuls nervos care intră în creier.
Înregistrările incorecte s-au efectuat în condiții de întuneric sub lumină roșie slabă la două nivele de iluminare pentru a descoperi răspunsurile combinate maxime ale tijei și ale conului. Datele au fost verificate pentru normalitate utilizând testul Kolomogorov-Smirnov. Toate datele nu au indicat nicio abatere de la distribuția normală.
Ce este distrofia retinei
Multe boli hepatice ale rinichilor au funcții retiniene care sunt diagnostice. Diagnosticul bolii renale ereditare este important din cauza riscului de complicații renale și sistemice, consecințe asupra altor membri ai familiei, predictibilității cursului clinic și posibilității tratamentului.
Boala este destul de obișnuită în timpul nostru. Această patologie afectează adesea vârstnicii și copiii.
Tipuri de distrofie retiniană

- Ereditar (primar):
- generalizată: degenerarea retiniană alb pigmentată și punctată, amaroza congenitală Leber, etc;
- centrală (vedere centrală afectată): boala Stargardt, boala Besta, degenerarea maculară senilă etc;
- periferică (tulburare de vedere laterală): retinoschiză juvenilă X-bolnavă, boala Wagner, boala Goldman-Favre etc.
- Obținut (secundar).
Distrofia secundară se dezvoltă pe fondul impactului factorilor externi sau interni asupra retinei, listați pe cele principale de mai jos.
În plus, anomaliile retiniane pot ajuta la explicarea patogenezei bolii renale și uneori pot fi folosite pentru monitorizarea cursului acesteia. Moștenirea bolii renale, care este prezentă atât la adulți, cât și la copii. Moștenirea bolii renale este suspectată în prezența unui istoric familial de simptome similare sau atunci când anomalii afectează mai multe sisteme fără altă explicație evidentă. Excepțiile cunoscute, bazate pe cunoștințele actuale, sunt forme autosomale dominante și recesive ale bolii polichistice a rinichilor, bolii renale musculare chistice și nefropatiei subtile a membranei bazale.
Factori predispozanți
- Intoxicația cu.
- Infecție.
- Tulburări circulatorii.
- Bolile sistemice ale țesutului conjunctiv.
- Endocrine patologice, inclusiv diabetul zaharat.
- Boli ale inimii și vaselor de sânge.
- Tulburări metabolice.
- Fumatul.
- Ochii răniți.
- Boala oculară (miopie, etc.).
- Sarcina.
- Obezitatea.
Explicații clinice ale bolii

Moștenirea bolii renale asociată cu pierderea auzului. Moștenirea renală moștenită include adesea retina, deoarece rinichii și retina se dezvoltă în aceeași etapă embrionară și împărtășesc căile de dezvoltare; barieră de filtrare glomerulară și tranziție retinochoid structural similară; glomerulul și corioretina sunt atât straturi mari de capilare, cât și celule epiteliale și epiteliale epiteliale ale pigmentului retinian, în mod critic dependent de funcționarea ciliei.
Modalități de dezvoltare a rinichiului și a ochilor
Celulele epiteliale au funcții specifice organelor, membranele de bază suportă structurile adiacente și formează o barieră care împiedică trecerea macromoleculelor, iar capilarele asigură hrană și elimină deșeurile din celule metabolice active din apropiere.
- Deteriorarea viziunii crepuscul.
- Fotofobie.
- Scaderea vederii centrale.
- Distorsiunea formei obiectelor.
- Apariția flash-urilor, zboară în fața ochilor.
- Scăderea acuității vizuale.
Degenerarea retiniană este o boală progresivă și în cele din urmă duce la o scădere treptată a vederii și orbire. Poate fi combinat cu nistagmus, strabism. Adesea are un curs asimptomatic. Cele mai nefavorabile în ceea ce privește prognosticul sunt degenerarea ereditară a retinei.
Mutațiile care afectează proteinele din cilia-podocite sau structurile asociate conduc la boala renală chistică, incluzând nefrofiză și variantele acesteia și sindromul Bardé-Bidel. Retina este de obicei afectată, iar alte caracteristici clinice includ pierderea auzului, dezvoltarea anormală a membrelor și a numărului, întârzierea dezvoltării, inversul sinusului, bolile hepatice și respiratorii, precum și infertilitatea.
Anomaliile de exfoliere a bolii renale ereditare includ colobom, peritoneu, atrofie și pigmentare, hamartom, anomalii vasculare și cristale. Aceste modificări pot fi, de asemenea, însoțite de asocierea reticulară a insuficienței renale, cum ar fi hemoragia, îngustarea arteriolelor, exsudatele, atacurile de cord, calcificarea și degenerarea maculară.
Luați în considerare cele mai frecvente opțiuni pentru distrofia retinei.
Distrofia pigmentului retinian
Aceasta este o patologie ereditară a retinei, în care bățurile sunt în primul rând afectate. Cu câțiva ani înainte de apariția modificărilor la fundus la pacienții cu adaptare întunecată este perturbată. Principalele manifestări ale bolii sunt hemeralopia (orbirea nocturnă) și îngustarea câmpului vizual. Leziunea este bilaterală, caracterizată prin depunerea pigmentului în fund, în timp ce vasele sunt înguste, distrugerea fibrelor nervoase optice se dezvoltă. Orbirea, de regulă, ajunge la 40 de ani.
Moștenirea bolii renale asociată cu tulburări retiniene. Tulburări retinale la boala renală ereditară. Hală optică. Viziunea era normală în acest ochi, dar coloboma este de obicei asimetrică. Anomalie a gloriei dimineții. Viziunea a fost spartă. Retina centrală normală. Retina centrală a fost, de asemenea, normală în ochii altora. Aceasta este retina periferică a feței a cărei fund este prezentată. Colostomia retinei a fost prezentă la nivelul periferiei pe o bază bilaterală. Retinopatia ușurează macula și este bilaterală.
Viziunea nu a fost afectată; Retinopatie periferică în sindromul Alport. Din nou, viziunea nu este afectată. Creierul macular foarte moale, larg răspândit, în glomerulonefrită membranoproliferativă. Forma târzie a bolii renale dense, cu o spălare intensă a laptelui renal, hemoragie și atrofie. Acești oameni trebuie evaluați și monitorizați de un oftalmolog datorită riscului de complicații și a posibilității de tratament. Atrofia retiniană în sindromul Joubert, o altă formă de nefronofos, demonstrată prin tomografie de coerență optică.
Amauroză congenitală Leber
Boala se manifestă de la naștere și are un curs sever. Copiii nu au o viziune centrală, pot să se nască orbi sau să-și piardă vederea înainte de vârsta de 10 ani. Examinarea a evidențiat strabismul, nistagmusul, diverse simptome neurologice, pierderea auzului și întârzierea dezvoltării neuropsihice. La examinare, fundul ochiului dezvăluie depuneri de pigment, un cap nervos optic palid.
Pacientul a avut inițial o vedere slabă, care a progresat. Atrofia corio-retinei cu sindromul Kearns Syrah. Retina este diluată și depigmentată. Viziunea a fost afectată. Hamartom în scleroza tuberculoasă. Tumorul alb cu hemoragie liniară subțire a fost prezent în mod unilateral. Această anomalie apare la majoritatea pacienților cu scleroză tuberculoasă și ar trebui să fie diferită de infarctul retinei datorat hipertensiunii, diabetului sau lupusului. Vase de tirbușon în sindromul Fabry.
Distrofie de tip punct
Acest lucru este asimptomatic, dar se poate produce o pierdere bruscă a vederii cu ocluzia arterei centrale. Ele trebuie diferențiate de modificările hipertensiunii. Sindromul Von Hippel Lindau. Vasele de broască țestoasă observate care duc la hemangioblastomul chiar în afara câmpului. Evidențele lipidelor maculare evidente sunt cauzate de scurgeri de la vase anormale. Aceste defecte sunt de obicei situate în partea inferioară a irisului, chorioretinului sau discului optic. Colobomul discului optic se găsește în refluxul vesicoureteral și în alte boli structurale ale tractului urinar.
Micul distrofie albă a retinei
Boala se dezvoltă în copilărie, are un caracter familial, are un curs progresiv lent. La pacienții cu crepuscul și orbirea nocturnă, s-a îngustat câmpul de vedere. Pe fondus au apărut leziuni albicioase, semne de atrofie optică.
Sindromul colobomului renal
Un colobom este de obicei asimetric, de exemplu, cu un ochi normal sau cu un defect slab într-un singur ochi și cu o anormalitate centrală gravă în cealaltă. În mod similar, severitatea unui defect variază între membrii familiei individuale. Nu există tratament și acestea pot fi complicate de glaucom, detașarea retinei și retinopatia seroasă centrală. Nouăzeci la sută dintre copii au un colobom cu disc optic, chorioretin sau iris, 20-40% au o anomalie structurală a tractului urinar, incluzând un rinichi solitar, hipoplazie renală, un rinichi duplex sau reflux vezicoureteral, iar 60% prezintă defecte cardiace.
Boala Stargardt (distrofie maculară)
Această patologie se manifestă la vârsta de 3-5 ani, ereditatea împovărată predispune la dezvoltarea ei. Copiii se plâng de vedere încețoșată, fotofobie. După câțiva ani de la debutul bolii, câmpurile vizuale centrale cad, iar acuitatea vizuală scade rapid. Examinarea a evidențiat modificări ale epiteliului pigmentar și atrofiei nervului optic.
Problemele de rinichi sunt adesea asociate cu paralizia facială ipsilaterală. Pierderea acestor gene cauzează anomalii oculare și urogenitale, respectiv. Diafragma este insuficientă, iar nervul optic și fosa hipoplastică. Aceste anomalii pot fi complicate de cornee fragile, de glaucom și de detașarea retinei. 50% dintre pacienți dezvoltă cancer renal.
Bolile distrofice ale retinei
Este probabil un subtip al sindromului Joubert cu afecțiuni hepatice. Pacienții pot avea semnul unui dinte molar, malformația midbrainului și a creierului din spate, observată, de asemenea, în sindromul Joubert și fibroza hepatică congenitală. Deși au chisturi renale medulare, ele nu dezvoltă neapărat insuficiență renală. Coloboma nervului optic, retard mental, nistagmus și ataxie congenitală.
Cea mai bună boală
Baza acestei patologii este degenerarea maculară. Se dezvoltă în copilărie și adolescență și se caracterizează printr-o scădere a acuității vizuale. Când oftalmoscopia a dezvăluit o leziune bilaterală a zonei de la fața locului sub formă de vatră galbenă.
Degenerarea maculei senile
Se dezvoltă, de regulă, la persoane mai în vârstă de 50 de ani. Suferă în cea mai mare parte sexul feminin. Procesul de degenerare continuă adesea neobservat. Boala începe cu o deteriorare a vederii la unul sau două ochi și este considerată prezbiopie. Odată cu progresia procesului degenerativ apare dispariția completă a vederii.
Sindromul Alport
Acestea sunt evidente cu oftalmoscopie fundus și fotografii, mai ales fără imagini roșii. Drusen cu un depozit dens este mare și moale și se află la nivelul maculei. Persoanele cu afecțiuni renale și atrofie retiniană se plâng mai întâi de vederea slabă pe timp de noapte și chiar de vizibilitatea tunelului. Inițial, anomalia se manifestă numai cu ajutorul unei electroretinograme, dar ulterior aspectul retinei este anormal cu paloare, pigmentare și spotare, arteriole și venule slăbite și vase vasculare evidente.
Moștenirea este recesivă autosomală. Tulburări rare asociate cu mutații recesive în genele care codifică alte proteine în cilia, corpurile bazale sau centrozomii celulelor epiteliale ale rinichilor și retinei. Uneori mutațiile afectează două gene diferite, iar severitatea bolii poate depinde, de asemenea, de gena modificatoare. Acest sindrom este caracterizat prin scleroză glomerulară focală clinică și segmentată, obezitate, cogniție afectată și polidactilă. Fenotipul variază în familii, iar persoanele nu pot fi prezente până în deceniul al cincilea.
X retinoschiză juvenilă cromozomială
Aceasta este o distrofie ereditară retinală, caracterizată prin disecția ei. Este mai frecvent la bărbați. Copiii au scăzut vederea, îndoială. Examinarea dezvăluie chisturi retiniene gigantice, înconjurate de pigmenți care se pot rupe.
Boala Wagner
Aceasta este o boală ereditară, a cărei manifestare este un grad ridicat de miopie. Adesea, pacienții cu 10-20 de ani au opacități ale lentilelor și o îngustare concentrică a câmpului vizual. Boala poate fi complicată de glaucomul sau detașarea retinei.
Retina este normală timpuriu, dar atrofia fără prea mult pigmentare are loc mai târziu. Anomaliile vizuale includ electroretinogram anormal, orbire nocturnă, defecte în câmpurile periferice și orbire la vârsta adultă. Transportatorii au probabil defecte minore ale funcției retiniene.
Alte caracteristici clinice includ fibroza hepatică, ataxia cerebellară și mișcările anormale ale ochiului, inclusiv nistagmusul rotativ, retardarea psihomotorie și întârzierea dezvoltării. Anomaliile de exfoliere includ pigmentarea retinitei și, eventual, colobomul chorioretinal. Ca și în cazul sindromului vârstnic Loken, pigmentarea retinită este adesea severă, iar electroretinograma este anormală într-o fază incipientă.
Boala Goldman-Favre
În această patologie, distrofia pigmentară este combinată cu o leziune vitroasă, retinoschisis. Manifestările bolii sunt vedere încețoșată, orbire nocturnă și adaptare întunecată la întuneric. La pacienți, poate fi detectată pierderea în formă de inel a câmpului vizual sau constricția concentrică.
Insuficiența renală, auzul slab și tonusul muscular redus apar, dar nu există anomalii ale retinei. Sindromul Meckel-Gruber poate fi cauzat de mutații în aceleași gene ca sindromul lui Joubert, dar este fatal la începutul vieții. Se caracterizează prin displazie chistică a rinichilor, fibroză hepatică, encefalocolor occipital și polidacil.
Sindromul Jun este caracterizat de boala renală chistică, piept lung, îngust, scurt limbic, brachidactil și alte defecte scheletice. Tulburările respiratorii pot fi severe. Retardarea atrofiei apare, dar ușoară. Ei au, de asemenea, pierderi de auz, disfuncție hepatică, cardiomiopatie avansată, pubertate întârziată, obezitate și diabet fără retard mental, polidactilă sau hipogonadism. Atrofia retiniană are loc cu particule mari de pigment, atenuare vasculară și lustruire a discului optic.
diagnosticare
 Oftalmoscopia permite medicului să examineze fundul ochiului.
Oftalmoscopia permite medicului să examineze fundul ochiului. Diagnosticul distrofiei retinei se bazează pe manifestările clinice, istoricul bolii, datele examinării și examinarea de către oftalmolog. Pacienții sunt deținute:
Sindromul Alagil
Prezentarea are loc din copilărie până la maturitate precoce. Pierderea auzului, cardiomiopatia și diabetul sunt, de asemenea, frecvente, dar caracteristicile clinice variază între familii. Atrofia retiniană apare la cel puțin jumătate dintre pacienți, dar electroretinograma este anormală la 90%.
Sindromul Kearns-Sayre
Începutul este de obicei de până la 20 de ani, dar uneori mai târziu. Sindromul Kearns-Sayre este asociat cu un defect al vaselor tubulare tubulare, precum și oftalmoplegia externă cronică progresivă, ptoza, pierderea auzului, anomalii ale conducerii cardiace, ataxie cerebelică și diabet. În ciuda atrofiei și pigmentării retinei, pierderea vizuală este de obicei ușoară.
- examen clinic general (sânge, urină, biochimie);
- oftalmoscopie (vă permite să examinați fundul);
- investigarea presiunii intraoculare;
- angiografia fluoresceinei retinei (studiul vaselor retiniene);
- echoftalmografia (metoda de studiere a structurilor globului ocular cu ultrasunete);
- entoptometrie (vă permite să obțineți o imagine completă a stării funcționale a retinei).
tratament
Terapia pentru procesele distrofice retiniene ar trebui să aibă o abordare integrată. Scopul său principal este de a opri progresia bolii și de a îmbunătăți viziunea cât mai mult posibil. Dacă procesul patologic care afectează retina este secundar, atunci boala care a condus la aceasta ar trebui tratată mai întâi.
Aceste tulburări includ neurofibromatoza, scleroza tuberculoasă, boala Lindau von Hippel și sindromul Sturge-Weber. Sindroamele von Hippel din Lindau și Sturge-Weber provoacă, de asemenea, anomalii vasculare și sunt discutate mai târziu. Hipertensiunea apare datorită stenozei arterei renale proximale, coarctării aortice sau femocromocitomului.
Tipuri de scleroză tuberculoasă 1 și 2
Anomaliile de exfoliere includ ischemia, hamartomul, incluzând hammarota retiniană combinată și epiteliul retinal retinal, gliomul optic și atrofia nervului optic, secundar presiunii asupra nervului optic. 20% dintre pacienți nu au nicio mutație. Acest lucru duce la hematurie și, uneori, la cancer. 30% dintre pacienți au chisturi renale. Coloboma și tumorile pleoapelor sunt rare. Majoritatea pacienților au cel puțin o retină sau gamma gama optică.
Terapia de droguri
- Steroizi metabolici (nerobol, retabolil).
- Anticoagulante (heparină, Fraxiparină).
- Dezagregant (aspirină, clopidogrel).
- Vasodilatatoare (acid nicotinic, drotaverin, komplamin).
- Medicamente care îmbunătățesc microcirculația (pentoxifilină, Cavinton).
- Vitamine (A, B, C, E).
- Emoxipină, taufon sub formă de picături pentru ochi.
Tratamentul oferă rezultate mai bune în stadiile incipiente ale bolii. Drogurile sunt utilizate atât pentru tratamentul local, cât și pentru tratamentul general.
Tratamentul chirurgical
Cu ineficiența tratamentului conservator, pentru a îmbunătăți circulația sângelui și nutriția țesuturilor, se efectuează revascularizarea retinei. Se utilizează transplantul de fibre musculare de la mușchii rectului ochiului în spațiul perichondral.
Anumite rezultate ale tratamentului pot fi obținute după coagularea foto și laser. În același timp, impactul este îndreptat spre țesuturile deteriorate, care sunt separate de cele sănătoase, ceea ce împiedică răspândirea procesului și împiedică detașarea retinei.
concluzie
Dezvoltarea proceselor distrofice în retina ochiului amenință pacientul cu pierderea completă a vederii fără posibilitatea recuperării acestuia. Aceasta se întâmplă de obicei în stadiile tardive ale bolii, în absența unei terapii adecvate. Boala reduce semnificativ calitatea vieții pacienților, iar orbirea duce la dizabilități. În prezența acestei patologii, este necesar să se înceapă tratamentul cât mai curând posibil, când nu s-au produs încă modificări ireversibile. Acest lucru face posibilă păstrarea vederii și suspendarea bolii.
NTV, un program "Fără prescripție medicală", problema "degenerării retinei":
Canalul TV "CTV.BY", programul "Bună ziua, Doctor", problema "distrofiei retinei - ce este":
Datorită dezvoltării tehnologiilor înalte, în prezent majoritatea bolilor oculare sunt tratabile. Cu toate acestea, în practica oftalmică, există încă patologii cu care este foarte dificilă, uneori imposibil de rezolvat. Cu astfel de boli grave cu un motiv bun pot fi atribuite distrofie pigmentară a retinei. O trăsătură distinctivă a acestei boli este un curs progresiv lung, alternând cu perioade de afectare vizuală accentuată și remisii neașteptate. De regulă, boala începe să se dezvolte în mod activ în timpul adolescenței, până la vârsta de 20 de ani poate fi deja diagnosticată clar, iar până la vârsta de 40-50 de ani, orbirea apare de obicei.
simptome
Un simptom tipic al acestei patologii este insuficiența vizuală severă, în special atunci când se trece de la lumina normală la o lumină mai slabă, de exemplu, la apus. Și în întuneric există "orbire de noapte". Din punct de vedere științific, această condiție se numește hemeralopie. La început, vederea periferică scade considerabil, apoi nervii optici atrofiază cu timpul, iar vederea centrală se îngustează concentric. Acesta este deja un predicator al orbirii totale.
Caracteristicile bolii
Dystrofia pigmentată (degenerare, retinită) a luat numele datorită modificărilor caracteristice care au apărut în fundus. Pe retină de-a lungul retinei (situată la nivelul locului fibrelor nervoase), vasele formau depuneri de pigment maroniu închis. Sunt niște corpuri osoase de diferite forme și mărimi, asemănătoare cu grămezi de cărbune fin. În timp, apare decolorarea celulelor epiteliului pigmentar. Drept rezultat, fundul ochiului strălucește și devine ca un mozaic, care este creat prin interconectarea densă a vaselor coroide portocalii-roșii, convergând în centrul ochiului.
Pe măsură ce boala progresează, numărul de organisme osoase pigmentare crește, zona lor de distribuție se extinde. Treptat, conglomeratele de mase pigmentare pun punct de densitate întreaga retină. Apoi, modificările distrofice captează partea centrală a ochiului. Vasele retiniene se îngustează foarte mult, devin filiform și capul nervului optic capătă o tentă de culoare palidă, cu atrofie ulterioară. Dystrofia pigmentată se dezvoltă adesea în ambii ochi, dar în cazuri rare poate afecta un ochi.
Cauzele dezvoltării
Originea (etiologia) distrofiei pigmentale nu a fost încă studiată. Oftalmologii exprimă opinii contradictorii. Unii cred că cauza acestei patologii este hipofuncția apendicelui creierului. Alții tind să provoace daune toxice organismului sau tulburării poliglaniare. Totuși, alții susțin că dezvoltarea bolii este o consecință a tulburărilor beriberi, endocrine sau genetice. Ipoteza este destul de stabilă, încât excesul de lumină iluminează o substanță specială în organele de viziune - rodopsina. Ca urmare, funcția de actualizare a fotoreceptorilor retinieni este frustrat. În acest caz, toți experții sunt de acord că cauza principală a bolii este de natură ereditară de familie.
tratament
Distrofia pigmentară este diagnosticată folosind cercetări electroretinografice. Din păcate, prognosticul este slab. Procesul patologic poate fi încetinit. Principala direcție a tratamentului este îmbunătățirea trofismului retinei, expansiunea vaselor, inclusiv a celor coroidale. În acest scop, se utilizează acid nicotinic, acetilcolină, precum și preparate pe bază de heparină. În general, lista de medicamente care încetinesc progresia distrofiei pigmentale este destul de extensivă. Medicii folosesc medicamente tiroidiene, testiculare, ovariene, steroizi metabolici (de exemplu, Nerobolil, Retabolil), prescrie terapie cu vitamine (cu excepția vitaminei A).
Informațiile de pe site-ul nostru sunt informative și educative. Cu toate acestea, această informație nu este în niciun caz un beneficiu auto-medicamente. Asigurați-vă că vă adresați medicului dumneavoastră.
- VKontakte 0
- Google+ 0
- în regulă 0
- Facebook 0